7.
Resolución estructural

En el
contexto de este capítulo Vd será invitado
también a visitar estos apartados...
La pregunta que
implícitamente nos
estamos
planteando desde los primeros
capítulos de estas páginas es:
¿Podemos
"ver"
la estructura interna de los cristales?,
es decir,
¿Podemos
"ver" los
átomos y las moléculas que están
dentro de los cristales?
Pues
bien, ¡ la
respuesta es sí !



Izquierda:
Estructura
molecular
de un enzima
presente en la superficie del neumococo
Centro:
Empaquetamiento
cristalino de una
molécula orgánica sencilla, mostrando la celdilla
elemental
Derecha:
Detalles
geométricos de la interacción entre
moléculas en un fragmento de una proteína
Y además, tal como muestran los ejemplos de arriba,
podemos
ver la estructura molecular por grande y compleja que ésta
sea
(figura de la izquierda), podemos ver el empaquetamiento de las
moléculas en la red cristalina (centro), y podemos ver
también
cualquier detalle geométrico e incluso los diferentes tipos
de
interacciones entre las moléculas o entre partes de ellas
(derecha).
Sin embargo, para que el
lector pueda comprender las bases sobre las que se fundamenta esta
respuesta, es necesario introducir o refrescar algunos conceptos que
nos
servirán para comprender el razonamiento de esta
afirmación...
En apartados anteriores hemos visto que
los cristales son la
manifestación por excelencia de la materia ordenada,
constituida
por asociaciones de átomos y/o
moléculas, que
corresponden a un estado natural de la misma, de mínima
energía.
Sabemos también que los cristales pueden
describirse
mediante unidades que se repiten en las tres direcciones del
espacio, y que a dicho espacio lo conocemos con el nombre de espacio
real ó directo. A esas unidades repetitivas las
denominamos celdillas
elementales (que además nos sirven como sistema
de referencia para describir
la posición de los átomos). Este espacio real ó directo
es el mismo en donde vivimos nosotros y se describe por la función de
densidad
electrónica, ρ(xyz),
definida en cada punto de coordenadas (xyz)
de la
celdilla, en donde, además, operan elementos
de simetría que repiten a los átomos
y moléculas en el interior de dicha celdilla.
 Celdilla elemental (izquierda) cuyo
apilamiento forma un cristal (derecha)
Celdilla elemental (izquierda) cuyo
apilamiento forma un cristal (derecha)
 Los
motivos (átomos, iones, o moléculas) se repiten
en el interior de la
celdilla elemental mediante las operaciones de simetría.
Las celdillas elementales se apilan
siguiendo las reglas de la red cristalina, formando un cristal .
Los
motivos (átomos, iones, o moléculas) se repiten
en el interior de la
celdilla elemental mediante las operaciones de simetría.
Las celdillas elementales se apilan
siguiendo las reglas de la red cristalina, formando un cristal .
También
hemos
aprendido que los rayos X
interaccionan con
los electrones de los átomos contenidos en los cristales,
dando lugar a un patrón de difracción,
también
conocido como espacio
recíproco, con las propiedades de un
retículo, con una cierta
simetría, y en el que también se
puede definir
una celdilla de repetición (celdilla
recíproca),
en cuyos nudos está almacenada la información
sobre la intensidad de la difracción.


Izquierda:
Interacción
entre las ondas
dispersadas por dos electrones. Las ondas resultantes muestran zonas
de oscuridad (interferencia destructiva), dependiendo del
ángulo de
dispersión. Animación originalmente tomada
de physics-animations.com.
Derecha: Una de
los cientos de imágenes
de un patrón de difracción de un cristal de
proteína. Los ennegrecimientos de la imagen son la
consecuencia de la
dispersión cooperativa (difracción) de los
innumerables
electrones contenidos en todos los átomos que constituyen el
cristal.
Las
ondas dispersadas de un modo
cooperativo, denominado difracción, se
componen a su
vez, en cascada, unas con otras, dando lugar a ondas totales
resultantes
en cada dirección de difracción, de tal modo que,
dependiendo del "desfase"
(el retraso o el adelanto) de unas con respecto a otras,
éstas
se
suman o se restan, tal como se muestra en las figuras de abajo:
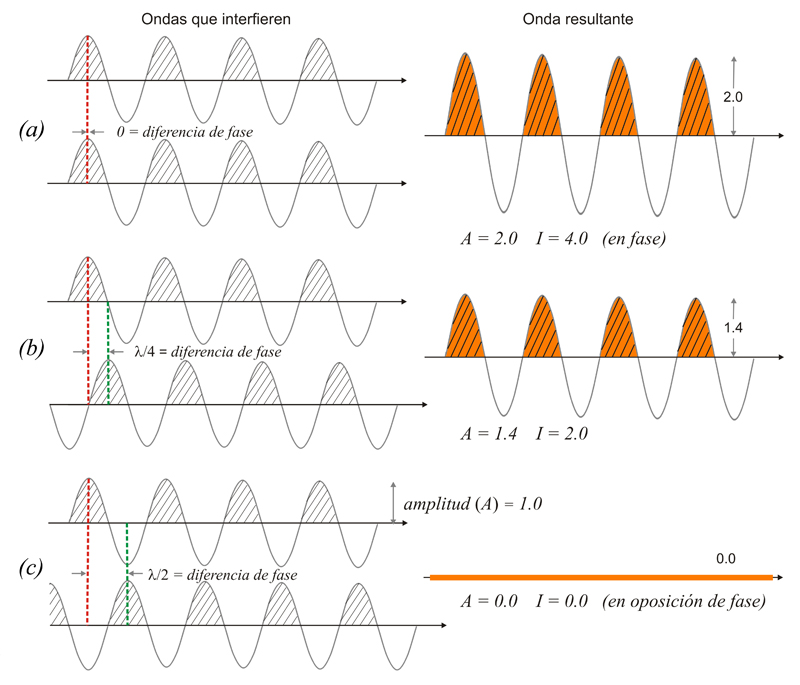 Composición
de dos ondas. A
= amplitud
resultante; I
=
intensidad resultante
(~ A2)
(a)
totalmente
en fase (el efecto total es
la suma de ambas)
(b)
con
un cierto desfase (hay suma pero no
total)
(c) en
oposición de fase (la
resultante es nula)
Composición
de dos ondas. A
= amplitud
resultante; I
=
intensidad resultante
(~ A2)
(a)
totalmente
en fase (el efecto total es
la suma de ambas)
(b)
con
un cierto desfase (hay suma pero no
total)
(c) en
oposición de fase (la
resultante es nula)
Entre los espacios
mencionados
(directo
y
recíproco)
existe una relación
holística (el
detalle de uno influye en el todo
del otro, y
el todo del uno influye en el detalle del otro). Esta
relación
es matemáticamente una transformada de Fourier que
no
podemos
resolver en sentido izquierdo del esquema de más abajo, ya
que el experimento de difracción no nos proporciona una de
las magnitudes fundamentales para resolver la ecuación, las fases
relativas (Φ)
de los haces de difraccion:


Izquierda:
Relación
holística
entre espacios directo (izquierda) y recíproco (derecha).
Cada
detalle del espacio directo (izquierda) depende de la totalidad de la
información existente en el espacio recíproco
(derecha),
y viceversa... Cada detalle del espacio recíproco (derecha)
depende de la
totalidad de la información del espacio directo (izquierda).
Derecha:
Representación
gráfica del desfase entre ondas.
Fase relativa de una onda respecto de otra
El esquema
de abajo, con ayuda del siguiente párrafo, resumen lo que
supone la
resolución de una estructura cristalina mediante la
difracción de rayos X...
Los átomos, iones y moléculas se empaquetan
ordenadamente en unidades (celdilla elemental) que se apilan
en tres dimensiones para formar un cristal en el espacio que
denominamos espacio directo o real. Los efectos de
difracción del cristal se pueden representar como puntos de
un espacio matemático reticular que denominamos red
reciproca. Las intensidades de difracción, es decir, los
ennegrecimientos de esos puntos de la red recíproca
representan los módulos de unas magnitudes vectoriales
fundamentales, que denominamos factores de estructura. Si llegamos a
conocer no sólo los módulos de dichos vectores
(las intensidades), sino sus orientaciones relativas (es decir, sus
fases relativas), seremos capaces de obtener el valor de la
función de densidad electrónica en cada punto de
la celdilla elemental, proporcionando así las posiciones de
los átomos que forman el cristal.
 Esquema sobre conceptos
básicos de la
cristalografía: espacios directo y recíproco. Se
pretende obtener información de la parte izquierda del
esquema (espacio directo) a partir del experimento de
difracción (espacio recíproco).
Esquema sobre conceptos
básicos de la
cristalografía: espacios directo y recíproco. Se
pretende obtener información de la parte izquierda del
esquema (espacio directo) a partir del experimento de
difracción (espacio recíproco).
LA
DENSIDAD ELECTRÓNICA
Conocer
(ó ver)
la estructura interna de un cristal supone poder resolver una
función matemática que define la denominada
"densidad
electrónica", que es una función que
está definida
en cada punto de la celdilla
unidad, ó celdilla elemental (un concepto
básico
de la estructura de
los
cristales y que se introducía en algún apartado
anterior).
Esta
función de densidad
electrónica,
representada por la letra griega ρ
tiene un
valor determinado en cada punto (x,
y,
z)
de la celdilla
elemental
y allí en donde toma valores máximos
(estimados en términos de "electrones por Angstrom
cúbico") es en donde estarán localizados los
átomos que componen un cristal, dándonos por lo
tanto la localización de los mismos.



Fórmula
1. Función
que define la densidad
electrónica en un
punto de coordenadas (x, y, z)
en el interior de la
celdilla elemental
- F(hkl)
representa las ondas resultantes de la dispersión
de todos los
átomos en cada una de las direcciones y se denominan factores
de estructura. Sus módulos están
directamente relacionados con las
intensidades de las reflexiones del patrón de
difracción.
- h,
k, l son
los
índices de Miller de las
reflexiones y Φ(hkl)
representa las
denominadas "fases" de las reflexiones (los desfases de unas ondas
respecto de otras). V
representa el
volumen de la celdilla elemental. En teoría el
sumatorio debería
extenderse desde -∞ hasta +∞,
pero como el
patrón de
difracción (= espacio recíproco, representado por
los factores de estructura) es finito, el resultado final del sumatorio
contiene pequeños errores de truncamiento.

 Izquierda: Aspecto
de una zona del mapa de densidad
electrónica de un cristal de proteína, antes de
su interpretación
Derecha: El mismo
mapa de densidad
electrónica de la izquierda interpretado en
términos de un fragmento peptídico
Izquierda: Aspecto
de una zona del mapa de densidad
electrónica de un cristal de proteína, antes de
su interpretación
Derecha: El mismo
mapa de densidad
electrónica de la izquierda interpretado en
términos de un fragmento peptídico
La ecuación de más arriba (Fórmula
1) representa
la transformada
de Fourier entre el espacio
real (en donde están los átomos)
representado por la función ρ y el espacio
recíproco (en donde está el
patrón de difracción) representado por los
factores de estructura y sus
fases. La Fórmula 1 demuestra
igualmente el llamado carácter "holísitico" de la
difracción, ya que para obtener el valor de la densidad
electrónica en un
sólo punto de coordenadas (xyz)
es necesario sumar las contribuciones
de todos los factores de estructura.
Los
factores de estructura F(hkl)
son ondas
y se
pueden representar también como vectores con sus
módulos, las amplitudes [F(hkl)],
y sus fases Φ(hkl) medidas
respecto de un origen
común de fases.
Cuando la celdilla elemental es
centrosimétrica, cada átomo de
coordenadas (xyz)
tiene otro idéntico
en coordenadas (-x,-y,-z), lo cual implica que se cumpla
la ley
de Friedel [F(h,k,l) = F(-h,-k,-l)]. Ello implica que la expresión de la
densidad electrónica (Fórmula
1) se simplifique,
convirtiéndose en la expresión de la Fórmula
1.1, y las fases de los factores
de estructura también se
simplifican, pues sólo podrán ser 0º
ó
180º...
Fórmula
1.1. Función
que define la densidad
electrónica en un
punto de coordenadas (x,
y, z)
en una celdilla elemental centrosimétrica.
Es importante darse cuenta que la cantidad y calidad de
información que proporciona la función de
densidad
electrónica, ρ,
es muy dependiente de la cantidad y calidad de los datos
que se suministren a la fórmula, que no son otros
que los
factores de estructura, F(hkl) (módulos y fases).
Más adelante veremos que los módulos se
obtienen directamente del experimento de difracción.
Si su navegador es compatible
con Java, como ejercicios visuales y
prácticos se recomienda visitar:
- esta
aplicación "applet" de Steffen Weber
que se abrirá en una nueva ventana. Escoja un color con el
ratón. Pinche un determinado número de veces
(tantas como
desee) sobre la ventana de la izquierda. Cambie de color si lo desea,
etc. y finalmente active la barra inferior derecha que aparece con el
nombre "Fourier transform". En la ventana de la derecha
aparecerá la transformada
de Fourier del conjunto de puntos de la izquierda,
- o, alternativamente esta otra
aplicación Java, algo
más completa, (original de Nicholas
Schöni y Gervais Chapui, Escuela
Politécnica Federal de Lausanne,
Suiza), que puede Vd. descargar (completamente libre de virus) desde el
enlace mostrado y ejecutar en su propio computador. Esta
aplicación calcula la transformada
de Fourier de
distribuciones bidimensionales de una función de densidad
electrónica ρ(x),
es decir, del espacio real, generando el espacio recíproco
correspondiente, es decir, amplitudes y fases de los factores de
estructura. Puede igualmente funcionar
a la inversa, es decir, generando la distribución ρ(x)
a partir del espacio recíproco. Esta aplicación
incorpora varias herramientas que pueden hacer comprender el diferente
papel que juegan las amplitudes de los factores de estructura (es decir, las intensidades de
difracción) y sus
correspondientes fases, e incorpora también la posibilidad
de simular una función
de Patterson.
La expresión
analítica de los factores
de estructura, F(hkl), es sencilla y en ella interviene una
nueva
magnitud (ƒj ) denominada
factor
de dispersión atómica (definida
en otro apartado anterior) que
da cuenta del poder con el que los electrones de los
átomos j
dispersan a los rayos X:
Fórmula
2. Factor de
estructura para cada haz
difractado. Esta función es la "transformada de Fourier" de
la
Fórmula
1.
Esta
expresión recoge las dispersiones f
de todos los
electrones de todos los átomos j
que
están contenidos en la celdilla elemental del cristal
Desde
el punto de vista experimental es relativamente sencillo poder medir
las amplitudes [F(hkl)] resultantes de todas las ondas
difractadas de un cristal. Basta para ello disponer de una fuente de
rayos X, de un cristal de la sustancia en estudio y de un detector
apropiado. En la
práctica se miden las
intensidades, I(hkl),
que están directamente relacionadas con las amplitudes a
través de:

Fórmula
3. Relación
entre el
módulo de los factores de estructura |F(hkl)| y la
intensidad I(hkl)
de los puntos del
patrón de difracción.
K es un factor que lleva los factores
de
estructura experimentales (Frel)
a la escala absoluta, es decir a la escala de
los factores de estructura calculados (teóricos). Este
factor se
puede determinar, de modo aproximado, usando los datos experimentales
mediante el denominado plot de Wilson.
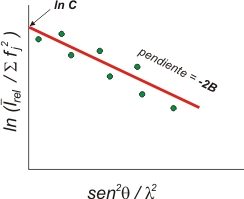 Plot
de Wilson
Plot
de Wilson
I
rel
representa la intensidad promedio (en escala relativa) en un
determinado intervalo de θ, fj es la
suma al cuadrado de los
factores atómicos de dispersión en esa zona
angular, y λ es la longitud de onda de los rayos X.
La
información aproximada que proporciona este
gráfico, en
el que se ha ajustado una línea recta a la
distribución
de puntos experimentales, está relacionada con:
- El valor de la
ordenada en el origen es el logaritmo neperiano de C,
relacionado con el factor de escala K
( = 1 / √C),
que
lleva los factores de estructura experimentales a una escala
próxima a la absoluta (a la de los factores de estructura
teóricos), es decir a la de aquellos que se
podrían calcular con el modelo
estructural.
- La pendiente de
la recta representa el valor de -2B,
en donde B
es un factor de vibración térmico,
isotrópico,
general para todos los átomos de la estructura.
A
es un factor de absorción, que puede estimarse
conociendo la composición del cristal y sus dimensiones.
L
es el denominado factor de Lorentz, responsable de
corregir la distinta
velocidad angular por la que pasan los puntos recíprocos por
la
superficie de la esfera de Ewald). Para geometrías con
goniómetros de cuatro círculos es un factor tan
sencillo como 1/sen
2θ, en donde θ
representa el ángulo de Bragg
de la reflexión (punto recíproco).
p
es el factor de polarización, que corrige el efecto de la
polarización del haz incidente sobre el cristal y viene dado
por la expresión (1+cos22θ)/2,
en donde igualmente θ
representa el ángulo de Bragg
de la reflexión (punto recíproco).
EL PROBLEMA DE LAS FASES
Sin embargo, para poder calcular la
función de densidad
electrónica
(ρ(xyz)
en Fórmula
1, más arriba), y por lo tanto
poder
saber la
localización de los átomos en el interior de la
celdilla,
necesitamos conocer también el desfase
entre las ondas (variable Φ(hkl)
en la
ecuación de
la densidad
electrónica) y esta información "se
nos escapa"
durante el
proceso de medida experimental, ya que no existen
técnicas
experimentales para medir esos desfases... El problema está, pues,
servido y esta dificultad es la que
ha dado nombre al concepto del problema
de las fases.
Esta circunstancia es también fácil de entender
si
comparamos el experimento de difracción (entendido como
procedimiento para ver las moléculas) con el microscopio
óptico convencional.
 Ilustración
sobre el problema de la fase. Comparación entre un
microscopio
óptico y la difracción de los rayos X en
términos
de lo que podría denominarse el "microscopio imposible" de
rayos X, ya que no existen lentes capaces de recombinar los rayos X.
Ilustración
sobre el problema de la fase. Comparación entre un
microscopio
óptico y la difracción de los rayos X en
términos
de lo que podría denominarse el "microscopio imposible" de
rayos X, ya que no existen lentes capaces de recombinar los rayos X.
En el microscopio
óptico convencional un foco de luz visible
ilumina la muestra y ésta dispersa los haces incidentes que
ruego se recombinan (con intensidades y fases) en un sistema de lentes
que dan lugar a la imagen ampliada del objeto en observación.
Pues bien, en lo que podemos denominar microscopio
imposible de rayos X, es decir en el proceso de visionado
del interior de los cristales (para determinar la estructura
de las moléculas), nuestra fuente de luz es especial (son
los rayos X, con longitudes de onda próximas al Angstrom) y
el cristal también dispersa (difracta) los haces. Sin
embargo, experimentalmente no disponemos de un sistema de lentes que
hagan el papel de recombinar de nuevo esos haces dispersados y nos
tenemos que contentar con "la fotografía" que
éstos nos dejan, por ejemplo, en una placa
fotográfica o en un detector. La señal medible
sobre el detector son las intensidades de los haces difractados, que
son proporcionales a las amplitudes [F(hkl)].
Pero sobre las fases, Φ(hkl),
nada podemos concluir y esta circunstancia nos impide la
resolución directa de la función de la densidad
electrónica mencionada en la ecuación del
principio de esta página.
Necesitamos, pues, métodos alternativos para recuperar esa
información perdida...
RESOLUCIÓN
DE ESTRUCTURAS
Conocido el problema
que se nos plantea tras obtener el patrón de
difracción (el
problema de la fase), veamos pues cuáles son
los pasos generales a los que se enfrenta un cristalógrafo
para ver
(resolver) la estructura de un cristal y la de las
moléculas, átomos o iones que contiene (ver
esquema de abajo)...

Esquema
general que ilustra el proceso de resolución de estructuras
moleculares
y cristalinas mediante la difracción de rayos X. Este
proceso
consta de diferentes pasos que han sido tratados
anteriormente, o
que se describen más abajo:
- Disponer u obtener un cristal
apropiado para
el experimento, con calidad y tamaño adecuados. Se
verá en otro apartado.
- Obtener el patrón de
difracción
con la longitud de onda adecuada. Se vió en otro
capítulo.
- Evaluar el patrón de
difracción
para obtener los parámetros
reticulares (celdilla
elemental), simetría (grupo
espacial) e intensidades
de difracción.
- Resolver la función
de densidad
electrónica, obteniendo previamente algún tipo de
información sobre las fases de los haces
difractados. Punto
clave de la resolución estructural, que se verá
más abajo.
- Construir un modelo estructural inicial,
interpretando la
función de la densidad electrónica y completar el
modelo
obteniendo las posiciones de los átomos restantes.
Se verá más abajo.
- Refinar
el modelo, ajustando todas las
posiciones
atómicas para conseguir que el patrón de
difracción calculado con dichas posiciones sea lo
más
parecido posible al patrón de difracción
experimental, y
finalmente validar y representar el modelo total obtenido.
Se
verá en otro capítulo.
Para que el estudio tenga
éxito hay que tener en cuenta algunos aspectos importantes,
tales como:
- La
sustancia en estudio debe ser pura para poder ser cristalizada (si
no lo está ya de forma natural, como en el caso de los
minerales).
- Los cristales pueden obtenerse mediante
técnicas muy variadas, desde las más simples como
evaporación o enfriamiento lentos, hasta otras
más complejas como difusión de vapor (o de
solvente), sublimación, convección, etc. Existe
mucha literatura accesible al respecto y el lector puede consultar, por
ejemplo,
las páginas del LEC,
Laboratorio de Estudios
Cristalográficos, para obtener
información adicional sobre técnicas
de cristalización específicas. En el caso de las
proteínas, el procedimiento más
extendido se basa en experimentos de difusión de
vapor, normalmente mediante las metodologías de
"gota
colgante" o "gota sentada" que puede consultarse en otro
apartado de estas páginas.
En este sentido, resulta muy relevante señalar los
últimos adelantos introducidos en el campo de la
nanocristalografía de rayos X en el tiempo de los
femtosegundos, que suponen un paso de gigante
para eliminar muchas de las
dificultades existentes en el proceso de la
cristalización, y en concreto para las proteínas (véase
el pequeño adelanto sobre los rayos X del laser de
electrones libres),
aunque quizá algo más tarde le interese leer con
detenimiento el capitulo completo dedicado a esta
novísima tecnología del XFEL.
- Si los cristales son adecuados se exponen
a los rayos X de acuerdo con alguna de las metodologías reseñadas
en otro
capítulo y se miden las intensidades
de difracción, lo que nos proporcionará a
través de ciertos cálculos sencillos, pero
cuidadosos
(evaluación de datos), las dimensiones de la celdilla
elemental, su simetría,
y los
módulos de los factores de estructura [F(hkl)]
a
partir de las intensidades. De todos estos aspectos, el más
dificultoso se refiere a la determinación de la
simetría
del cristal, ya que el conocimiento de ésta es crucial para
llevar a buen término la resolución de la
estructura.
Para ello, y tal como parece obvio, el cristalógrafo no hace
uso
del estudio visual del cristal, sino del propio patrón de
difracción, tal como se indica en un apartado
específico referente a la simetría de dicho
patrón y que invitamos a consultar.
- Es entonces cuando el
cristalógrafo se plantea la resolución de las
fases Φ(hkl)
mediante
diferentes métodos...
- Si las fases son
correctas, la función de densidad electrónica ρ(xyz)
mostrará
una distribución de máximos interpretable y
compatible con una estructura con sentido esteroquímico, y a
partir de ese momento sólo restarán algunos pasos
adicionales (construcción detallada del
modelo, ajuste
matemático y validación
estereoquímica) que nos
conducirán
al denominado modelo final de la estructura.
Pero veamos lo más
importante,
¿cómo
se resuelve el problema de las fases?...
LA FUNCIÓN DE PATTERSON
 Históricamente
hablando, la primera solución al
problema de las fases vino de la mano de Arthur
Lindo Patterson (1902-1966).
Históricamente
hablando, la primera solución al
problema de las fases vino de la mano de Arthur
Lindo Patterson (1902-1966).
Basándose
en la imposibilidad de resolver de un modo directo la
función de la densidad electrónica
(Fórmula 1, más arriba, o más abajo),
y
tras su aprendizaje sobre convolución de transformadas de
Fourier con el matemático estadounidense Norbert
Wiener, en 1934 Patterson
introdujo
una
nueva
función P(uvw)
(Fórmula
4, más abajo). Esta nueva
función, que define en un nuevo espacio (espacio
de Patterson), puede considerarse sin
exageración como el desarrollo
singular más importante para la
Cristalografía,
tras el
propio descubrimiento de los rayos X por Röntgen
en 1895.
Su elegante fórmula, conocida como la
función de Patterson
(Fórmula
4, más abajo),
supone una simplificación
de la información contenida en la función de
densidad electrónica, ya que suprime la
información
de las fases, y los
módulos de los factores de estructura se
sustituyen por sus cuadrados. Es, pues, una función
que
puede calcularse de inmediato a partir de la información
experimental de que se dispone (las intensidades, que a su vez se
derivan de los módulos de los factores de estructura).
Formalmente, desde el punto de vista matemático, la
función de Patterson es equivalente a la convolución
de la función de la densidad
electrónica (Fórmula
1, más abajo) con
su inversa: ρ(x,y,z)
* ρ(-x,-y,-z).
Formula
1.
Función de densidad
electrónica calculada en un punto de coordenadas (x,y,z).
 Formula
4. Función
de Patterson calculada en un punto de coordenadas (u, v,
w). Esta
fórmula es una simplificación de la
Fórmula 1,
ya que el sumatorio se
realiza
sobre F2(hkl) y se asume que todas las
fases son cero.
Formula
4. Función
de Patterson calculada en un punto de coordenadas (u, v,
w). Esta
fórmula es una simplificación de la
Fórmula 1,
ya que el sumatorio se
realiza
sobre F2(hkl) y se asume que todas las
fases son cero.
Parece obvio que por el hecho de
haber
prescindido de la información crucial que contienen las
fases [Φ(hkl) en Formula
1],
la función de Patterson ya no mostrará
directamente la
posición de los átomos, tal como lo
haría la
función de densidad electrónica. En efecto,la
información que proporciona
la función de
Patterson sólo es un mapa
de vectores de posición entre átomos
(posiciones relativas).
Los
máximos de la función de Patterson son tanto
más altos cuanto mayores
sean los números de electrones de los átomos
implicados, lo cual supone
una gran ventaja en el caso de moléculas que contengan
especies de
número atómico alto. Calculada dicha la función, P(uvw), se trata pues de
interpretarla correctamente (obtener la posición absoluta de
algunos átomos) para, indirectamente, alcanzar el
conocimiento necesario,
fases Φ(hkl), que nos permitan
visualizar la función de la densidad
electrónica ρ(xyz),
pero esto será objeto de otro apartado
específico que
invitamos a visitar.
LOS
MÉTODOS DIRECTOS
El problema de la fase para cristales formados por moléculas
de tamaños pequeño y medio fue
resuelto muy satisfactoriamente mediante los llamados métodos
directos, gracias a varios autores a lo largo del siglo
XX, entre los que cabe especialmente mencionar a Jerome
Karle (1918-2013) y Herbert
A. Hauptmann (1917-2011), quienes compartieron
el
Premio Nobel de
Química en 1985 (sin olvidar el papel de Isabella Karle,1921-2017).
El hecho de que la densidad
electrónica deba ser cero o positiva en cualquier punto de
la celdilla cristalina, y la atomicidad de las moléculas,
genera ciertas limitaciones en la distribución de fases
asociada a los factores de estructura. En dicho contexto, los métodos
directos están basados en el establecimiento de
sistemas de ecuaciones que usan las intensidades de los haces
difractados y que describen dichas limitaciones. La
resolución de dichos sistemas de ecuaciones proporciona
información
directa sobre la distribución de
fases. Sin embargo, puesto que la validez de cada una de
estas
ecuaciones se establece en términos
probabilísticos, es necesario disponer de un gran
número de ecuaciones que sobredeterminen los valores de las
incógnitas (fases Φ(hkl)).

Se
trata de usar ecuaciones
que relacionan la fase de una reflexión (hkl) con
las de otras
reflexiones vecinas (h',k',l')
y (h-h' ,k-k', l-l'),
cuyas fases son probablemente
ciertas (P)...

en
donde Ehkl,
Eh´k´l´
y Eh-h',k-k',l-l' son los denominados "factores
de estructura normalizados", es
decir, factores de estructura corregidos por las vibraciones
térmicas de los átomos, puestos en la llamada
escala
absoluta y suponiendo que los átomos son puntuales. En otras
palabras, la "normalización" de los factores de estructura
convierte los valores |F|
medidos, en coeficientes (conocidos
como valores |E|) supuestamente producidos por
átomos puntuales en reposo.
Esta
metodología se denomina
"directa" por el hecho de que, en contraste con otros
métodos,
pretende obtener la información estructural (densidad
electrónica) de un modo relativamente directo, es decir,
obteniendo las fases de
los
factores de estructura directamente a partir de sus
módulos. Este procedimiento se
aplica con excelentes resultados para cristales y moléculas
de tamaños pequeño y medio, es decir, hasta un
centenar de átomos aproximadamente.
Invitamos al lector a visitar una excelente introducción a
los métodos directos que encontrará en este enlace
que ofrece la Unión Internacional de
Cristalografía.
MÉTODOS DE RESOLUCIÓN ESTRUCTURAL PARA
MACROMOLÉCULAS
En cristales que contienen moléculas
grandes, proteínas o enzimas, el problema
de la
fase puede resolverse mediante tres métodos,
dependiendo del caso:
(i)
introduciendo átomos altamente dispersores, o
método del Reemplazo Isomorfo Múltiple (MIR,
del
inglés, Multiple
Isomorphous
Replacement),
y por lo tanto basado en el método de Patterson;
(ii)
introduciendo átomos dispersores anómalos, o
método de Difracción Anómala
Múltiple (MAD,
del inglés Multi-wavelength
Anomalous
Diffraction),
y
(iii)
mediante el Reemplazo Molecular (MR,
del inglés Molecular
Replacement),
haciendo uso de un modelo estructural de una
proteína homóloga, previamente determinada.
EL
MÉTODO MIR (Multiple
Isomorphous
Replacement,
reemplazo isomorfo múltiple)
Este método, basado en la metodología de
Patterson, fue introducido por David
Harker, aunque los primeros en usarlo con éxito
fueron Max
F. Perutz y John
C. Kendrew,
galardonados con el Premio Nobel de Química en 1962, al
resolver por primera vez la estructura de una proteína, la
hemoglobina.
El método MIR
consiste en introducir, en la celdilla
cristalina,
átomos que sean grandes dispersores de los rayos X
(átomos con números atómicos
elevados), pero la dificultad estriba en el hecho de que los
átomos introducidos no deben distorsionar
la estructura
cristalina de la proteína nativa, es decir, que los
cristales así obtenidos deben ser isomorfos de los de la
proteína nativa.
En la práctica, este reemplazo
isomorfo se lleva a cabo difundiendo complejos de metales "pesados" a
través de los canales que poseen los cristales de
proteína, y en donde normalmente existen cadenas laterales
de aminoácidos con capacidad de coordinar a los
átomos metálicos (por ejemplo grupos SH de las
cisteínas). En el caso de las metaloproteínas es
posible reemplazar sus metales endógenos por otros
más pesados (por ejemplo Zn por Hg, Ca por Sm, etc.).
Los átomos "pesados" (al disponer de un gran
número de electrones) contienen un poder de
dispersión mayor que el de los átomos normales de
las proteínas (C, H, N,
O y S), y por lo tanto pueden llegar a modificar sensiblemente
la
intensidad del patrón de difracción del cristal
derivatizado frente al nativo. Son precisamente estas diferencias de
intensidad entre ambos patrones de difracción las que se
usan para calcular
un mapa
de vectores de situación relativa entre los
átomos pesados (mapa de Patterson), a
partir del cual es
relativamente sencillo determinar sus posiciones dentro de la celdilla
elemental.

Esquema gráfico de la función de Patterson
derivada de un cristal con tres átomos. Para obtener
gráficamente esta función a partir de la
estructura conocida de un cristal (figura izquierda) se trazan todos
los vectores posibles entre cada pareja de átomos (figura
central) y se trasladan equipolentemente (paralelos a sí
mismos) al origen de la celdilla de la función de Patterson
(figura derecha). En el extremo de dichos vectores se dan los valores
máximos de la función de Patterson, cuyas alturas
son proporcionales al producto de los números
atómicos de los átomos implicados. Las posiciones
de estos máximos (coordenadas u, v, w) representan las
diferencias de coordenadas entre cada pareja de átomos del
cristal, es decir: u=x1-x2, v=y1-y2,
w=z1-z2.
Conocidas
las posiciones de los
átomos pesados, se
calculan ahora los factores
de estructura mediante la Fórmula 2 (ver esquema
de más abajo),
sus módulos |Fc(hkl)|
y sus fases Φc(hkl),
en donde el subíndice c
significa "calculado". Mediante la Fórmula 1 se
calcula ahora un mapa de densidad electrónica,
ρ(xyz),
usando los módulos de los factores de
estructura observados en el experimento,
|Fo(hkl)|
(que contienen la
contribución de la estructura total) y
combinándolos con las fases calculadas, Φc(hkl).
Si
el valor de estas fases calculadas es suficientemente aproximado, el
mapa de densidad electrónica calculado contendrá
información suficiente para su interpretación
posterior, añadiendo así información
adicional (más posiciones atómicas) al modelo
estructural (ver esquema de más abajo).
En definitiva, los pasos a seguir en el método MIR son:
- Preparación de uno, o varios derivados de
átomo pesado del cristal de la proteína. Un
primer control del isomorfismo viene dado por la comparación
de las dimensiones de las celdillas del cristal nativo y del derivado.
- Toma de datos de difracción para los cristales
de proteína nativa y de derivados.
- Aplicación de la función de Patterson
para la determinación de las coordenadas de los
átomos pesados.
- Refinamiento de los parámetros de los
átomos pesados y cálculo de las fases de los
haces difractados de la proteína.
- Cálculo de la densidad electrónica de
la proteína.
EL
MÉTODO MAD (Multi-wavelength
Anomalous
Diffraction,
difracción anómala múltiple)
Los cambios que se provocan en la intensidad de la
difracción al introducir átomos pesados en los
cristales de proteína se pueden considerar como
modificaciones químicas de la difracción. De modo
análogo, se pueden provocar cambios en las intensidades de
la difracción modificando las propiedades físicas
de los átomos. De este modo, si la radiación X
incidente tiene una frecuencia próxima a la frecuencia
natural de oscilación de los electrones de un determinado
átomo, se produce la denominada dispersión
anómala, que modifica el factor de dispersión
atómico, ƒj (véase Fórmula 2),
de tal modo que su expresión se ve modificada con dos
términos, ƒ' y ƒ'', que dan cuenta de las componentes real
e imaginaria, respectivamente, de la fracción
anómala del factor de dispersión. Para los
átomos que se comporten anómalamente, su factor
de dispersión vendrá dado por la
expresión que se muestra en la Fórmula 5.

Fórmula
5. En presencia de
dispersión
anómala, el factor de dispersión
atómico, ƒ0
,
se ve modificado por dos términos adicionales que dan cuenta
de las partes real e imaginaria de la dispersión
anómala.
El lector interesado
en comprender el significado de la Fórmula
5 debería
consultar
el apartado específico referente al fenómeno de
la dispersión anómala.
Las variaciones de ƒ' y ƒ'' frente a la energía de los
rayos X se pueden calcular usando consideraciones teóricas,
tal como se muestra en la figura de abajo para el caso del cobre.
 Variación de las componentes
real e imaginaria del factor de dispersión
atómico del selenio
en función de la
energía de los rayos X incidentes. La línea
vertical señala la longitud de onda del CuKα.
Variación de las componentes
real e imaginaria del factor de dispersión
atómico del selenio
en función de la
energía de los rayos X incidentes. La línea
vertical señala la longitud de onda del CuKα.
Para
los valores de la energía
de los rayos X en los que existe resonancia, el valor de ƒ''
aumenta drásticamente, y al mismo tiempo el valor
de ƒ'
decrece. Este
hecho tiene gran importancia práctica si se tiene en cuenta
que muchos
de los átomos pesados usados en Cristalografía
tienen picos de
absorción a energías (longitudes de onda) que se
pueden obtener
fácilmente mediante radiación
sincrotrón. Así pues, los datos de
difracción recogidos en estas condiciones tendrán
una componente
normal, debida fundamentalmente a los átomos ligeros
(nitrógeno,
carbono e hidrógeno), y una componente anómala,
la cual será debida a
los átomos pesados, que producirá una
alteración en la fase global de
cada reflexión. Todo esto se traducirá en un
cambio de intensidad entre
las llamadas reflexiones de Friedel (parejas de reflexiones que en
condiciones normales deberían tener la misma amplitud e
idénticas
fases, pero con signos opuestos). El cambio de intensidad detectable
entre las parejas de Friedel recibe el nombre de difracción
anómala.
El método
MAD,
desarrollado
por Hendrickson y Kahn, implica
la medida de los datos de difracción de un cristal de la
proteína (que contenga un dispersor anómalo
fuerte) usando radiaciones de distintas energías: la que
maximiza ƒ'', la que minimiza ƒ' y una energía lejana de estas
dos. Combinando estos conjuntos de datos de difracción, y en
concreto analizando las diferencias entre ellos, es posible calcular la
distribución de amplitudes y fases que generan los
dispersores anómalos. El uso posterior de las fases
generadas por estos dispersores anómalos, como una primera
aproximación a las fases globales, permite calcular la
densidad electrónica para toda la proteína.
En general, en la práctica no es necesario introducir
átomos aislados, como dispersores anómalos, en el
cristal de proteína. Hoy en día es relativamente
sencillo obtener proteínas recombinantes en las que los
residuos de metionina están reemplazados por
selenio-metionina. El selenio, e incluso el azufre de la metionina, o
de la cisteína, son dispersores anómalos
adecuados para llevar a cabo un experimento MAD.
Las ventajas que
presenta el
método MAD
respecto del método MIR
son:
- Como los datos en la
técnica MAD
se recogen con un único cristal, desaparecen los
problemas de falta de isomorfismo tan habituales en la
técnica MIR.
- Mientras que el
factor de
dispersión atómico en ausencia de
dispersión anómala (ƒ0) decrece drásticamente con el
ángulo de dispersión, la componente
anómala del factor de dispersión
atómico (ƒ' + iƒ'' ) es independiente de dicho
ángulo, por lo que esta señal relativa aumenta
con la resolución del patrón de
difracción, es decir, a
ángulos de dispersión elevados. De este modo, las
estimaciones de fases mediante este método son, en general,
mejores a alta resolución. En el caso MIR, la falta de
isomorfía es más acusada en las reflexiones de
alto ángulo, por lo que en la mayor parte de los casos la
información de fases mediante esta técnica no se
puede obtener a partir de las reflexiones de resolución
mayor de unos 3.5 Å.
 Diagrama
de Argand mostrando la contribución a la
dispersión de un átomo dispersor
anómalo en
una matriz de dispersores normales, lo que implica la ruptura de la ley
de Friedel. Imagen tomada de los apuntes de
"Crystallography
101".
Diagrama
de Argand mostrando la contribución a la
dispersión de un átomo dispersor
anómalo en
una matriz de dispersores normales, lo que implica la ruptura de la ley
de Friedel. Imagen tomada de los apuntes de
"Crystallography
101".
- Fp
representa la contribución al factor
de
estructura de
los átomos dispersores normales para una
determinada
reflexión de índices hkl.
- Fa
representa la parte real (ƒ0
+ ƒ ' ) del factor de
dispersión de
los átomos dispersores anómalos.
- Fa''
representa
la parte imaginaria (ƒ '' ) del factor
de
dispersión de
los átomos dispersores anómalos.
- -Fp,
-Fa
y -Fa"
representan lo mismo que Fp, Fa
y Fa'', pero
para la
reflexión de índices -h, -k,
-l.
Cuando se dispone de
un modelo
estructural de una proteína con una secuencia de
aminoácidos homóloga, el problema de la fase se
puede resolver mediante la técnica del reemplazo molecular
(MR). La estructura de esta proteína homóloga se
considera como si fuera la proteína que va a determinarse y
sirve como un primer modelo que posteriormente será
refinado. Este procedimiento está basado en la
observación de que proteínas homólogas
en su secuencia peptídica, muestran un plegamiento muy
similar. El problema en este caso, consiste en transferir la estructura
molecular de la proteína conocida, desde su propio
empaquetamiento cristalino hasta el cristal de la proteína
con estructura desconocida. El posicionamiento de la
molécula conocida en la celdilla unidad de la
proteína desconocida, requiere determinar su correcta
orientación y su posición precisa. Ambas
operaciones, rotación y translación, se calculan
mediante las funciones denominadas de rotación y de
translación (figura de más abajo).
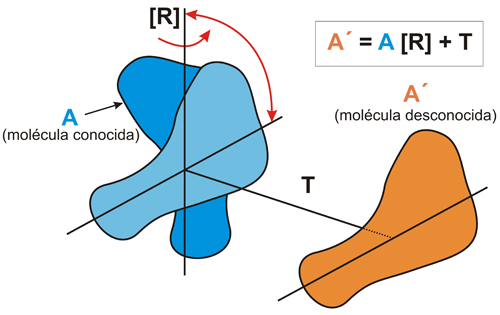 Esquema
del proceso de reemplazo
molecular (MR).
Esquema
del proceso de reemplazo
molecular (MR).
La
molécula de estructura conocida (A)
se gira mediante la operación de rotación [R]
y se traslada mediante T,
para llevarla a la posición que ocupa la molécula
problema (A’).
La
función de rotación. Si consideramos
el caso de dos moléculas idénticas, orientadas de
modo diferente, entonces la función de Patterson
estará compuesta por tres conjuntos de vectores. El primero
estará formado por los vectores de Patterson de una de las
moléculas, es decir, por todos los vectores
interatómicos dentro de una de ellas (también
llamados autovectores). El segundo será el correspondiente a
los vectores de la segunda molécula, que serán
idénticos a los de la primera molécula, pero
rotados debido a su distinta orientación. El tercer conjunto
de vectores lo constituirán los vectores cruzados que
describen los vectores interatómicos entre las dos
moléculas. Mientras que los autovectores estarán
confinados al volumen ocupado por la molécula, los vectores
cruzados se extenderán más allá de
este límite. La función de rotación R(α,β,γ)
intenta superponer los vectores de Patterson de una de las
moléculas con los de la otra hasta que se obtenga un buen
acuerdo. Esta metodología fue descrita por primera vez por
Rossman y
Blow.
R(α,β,γ)
= ∫u
P1(u) x P2(ur)
du
Fórmula
6. Función
de
rotación
P1
es la función de Patterson y P2
la función de
Patterson rotada, siendo u
el volumen del mapa de Patterson, en donde se
calculan los vectores.
La calidad de las soluciones de estas funciones se expresa mediante los
coeficientes de correlación entre las funciones de Patterson
experimental y calculada con la proteína conocida. Un
coeficiente de correlación alto entre dichas funciones
equivale a un buen acuerdo entre el patrón de
difracción
experimental y el calculado con la proteína conocida. Una
vez orientada y trasladada convenientemente la molécula
conocida, se calcula un mapa de densidad electrónica usando
los factores de estructura experimentales. Merece la pena consutar el magnífico
artículo que, sobre esta metodología,
publicó Eleanor Dodson.
Quizá el lector avanzado desee consultar
un magnífico artículo
que, a pesar de haber sido publicado en 2010, no ha perdido su vigencia
en relación con la descripción de las distintas
metodologías para la determinación de las fases
relativas
de los haces de difracción.
COMPLETANDO LA ESTRUCTURA
Todos estos métodos (Patterson, métodos directos,
MIR, MAD, MR)
proporcionan (directa o indirectamente)
un conocimiento aproximado de las fases, lo que denominamos fases
iniciales, que hay que mejorar. Tal como se indica en el
párrafo anterior, estas fases
iniciales calculadas, Φc(hkl), junto
con las amplitudes
experimentales observadas, |Fo(hkl)|,
nos permitirán calcular una
función de densidad electrónica,
también aproximada, y
sobre la cual podremos construir
nuestro modelo estructural. El proceso general a seguir se resume,
cíclicamente, en el esquema siguiente.
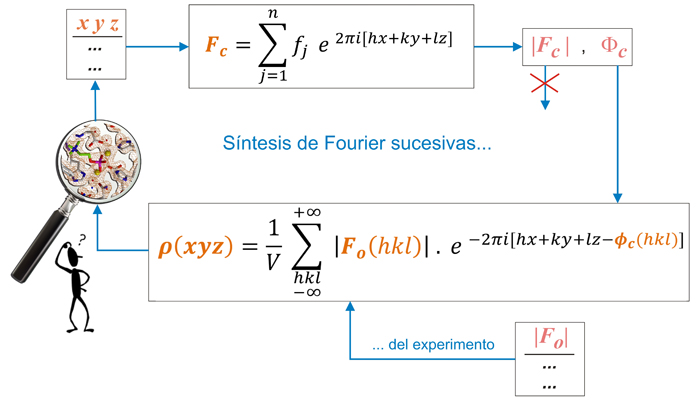
Las fases iniciales, Φc(hkl), se pueden combinar con los
módulos de los factores de estructura experimentales
(observados) |Fo(hkl)|
para calcular la función de densidad electrónica
(parte
inferior del esquema). Alternativamente, si el conocimiento inicial han
sido las posiciones atómicas (xyz)
de algunos átomos, éstos
proporcionarán las fases iniciales (parte superior del
esquema).
Esquema de los cálculos
sucesivos de un mapa
de densidad electrónica, ρ(xyz).
A partir de las
posiciones
atómicas conocidas (xyz)
se pueden obtener los módulos de factores de estructura y
fases calculados, |Fc(hkl)|
y Φc (hkl)
(parte superior). Los módulos de los factores de estructura
calculados son rechazados por estar evaluados a partir de una
estructura
parcial, mientras que se aceptan los módulos
experimentales, ya que son consecuencia de la estructura
real y completa. Así pues, la
función de densidad
electrónica se calcula con los módulos
experimentales de los factores de estructura, es decir,
los observados, |Fo(hkl)|, y se combinan con las
fases
calculadas a paritir del modelo parcial que se tenga, Φc(hkl).
La función de densidad electrónica así
calculada se evalúa cuidadosamente para
detectar la
posible
información añadida que se detecte, es
decir, posiciones de nuevos átomos. Y el
ciclo se
repite hasta que no se
obtenga información adicional. Históricamente
este
proceso se conoció como "síntesis sucesivas de
Fourier"
ya que la función de densidad electrónica, que en
teoría es una integral, se calcula como una suma de
Fourier.
De
un modo u otro (sea a partir de posiciones atómicas o
directamente desde las fases), si dicha información es
correcta,
la
función de densidad
electrónica así calculada
será interpretable y contendrá
información
adicional (nuevas coordenadas atómicas) que
podrán ser inyectadas en
el
ciclo del esquema superior hasta que se haya completado la estructura,
es decir
hasta que la última función ρ(xyz) no muestre modificaciones
respecto del último cálculo.
Los átomos más ligeros de la estructura (los de
menor número atómico, es decir, normalmente los
átomos de hidrógeno) son los más
difíciles de encontrar en un mapa de densidad
electrónica porque su factor de dispersión queda
prácticamente oscurecido por la dispersión de los
restantes átomos. Por esta razón, la
localización de los átomos de H se realiza por
medio de una función de densidad electrónica algo
modificada ("función de diferencias"), tomando como
coeficientes las diferencias entre los factores de estructura
observados y los calculados con el modelo conocido:

Fórmula
7. Función
de
densidad electrónica "diferencia"
En la práctica, si el modelo estructural obtenido es bueno y
el experimento dió lugar a factores de estructura precisos y
no existen errores tales como absorción de los rayos X por
el cristal, el mapa de Δρ proporciona
suficiente señal para localizar a los átomos de
H. Adicionalmente, para conseguir mayor señal procedente de
la
dispersión de estos átomos ligeros, esta
función se suele calcular exclusivamente con los factores de
estructura de menor ángulo, normalmente con aquellos que
aparecen a un valor de sen
θ / λ < 0.4
Siguiente
capítulo: El modelo estructural
Tabla de
contenido